Biology (SJCR)
Stechnolock Journal of Case Reports
Full Text
Volume 1, Issue 1
Giant Cervical Neurofibroma: A Case Report
*Corresponding Author: Mohamed Afellah, Otorhinolaryngology/ Head and Neck Surgery Department, Hassan II University Hospital, Sidi Mohamed Ben Abdellah University, Morocco, Tel: +212658887480, E-mail: Mohamed.afellah@gmail.com
doi: /sjcr.2021.1.106
Citation: Mohamed Afellah (2021) Giant Cervical Neurofibroma: A Case Report. Stechnolock J Case Rep 1: 1-8
Copyright: © 2021 Mohamed Afellah. This is an open-access article distributed under the terms of Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
The Von Recklinghausen disease or neurofibromatosis type 1 (NF1) is an exceptional autosomal dominant multisystem disorder.
This disease manifests by cutaneous (café-au-lait spots) and neurological (neurofibromas) symptoms. A malignant progression is rare but justify a long-term observation and multidisciplinary management.
We report in this study a 55-year-old female patient who consulted in surgical department for a swelling in her left part of the neck revealing a Von Recklinghausen disease.
Keywords: Neurofibromatosis, Neurofibroma, RMI, Immunohistochemistry
Introduction
Neurofibromatosis type 1 (NF1) or Von Recklinghausen disease is an autosomal dominant multisystem disorder with a nearly even split between spontaneous and inherited mutations.
Clinically, the dermic café-au-lait macula and tumors located along the nerves, called neurofibromas are classic manifestation. Complications depend on size, number and localization of these neurofibromas.
The diagnosis of NF1 is based on the clinic.
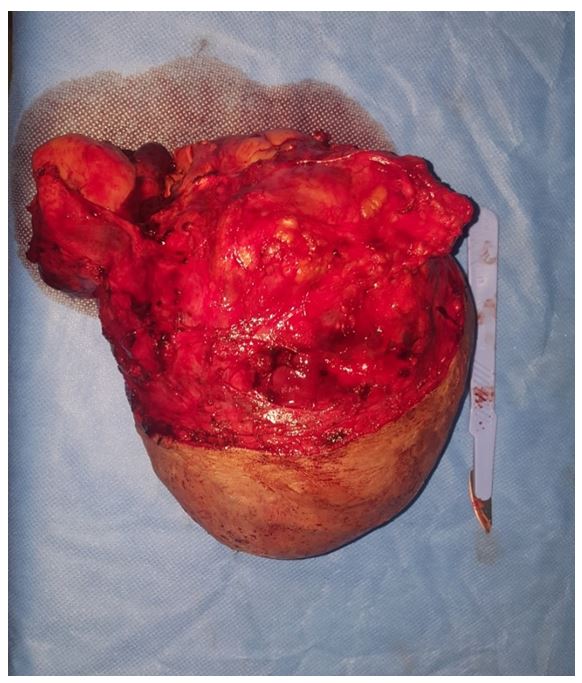
We report the case of a 55-year-old woman who consults for a right cervical mass measuring 16 cm revealing a Von Recklinghausen disease.
Observation
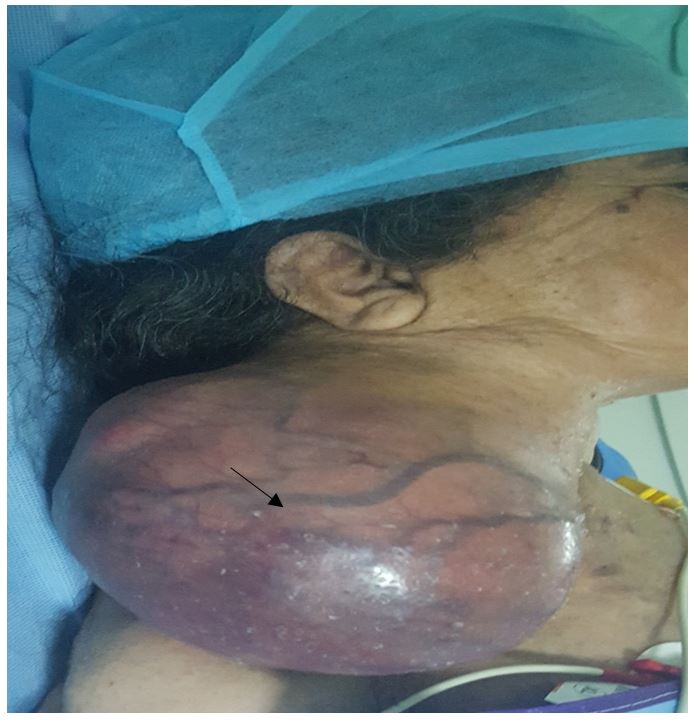
A 55-year-old female patient,with history of a toxic multi-nodular goiter taking anti-thyroid synthetics, presented a rapidly growing swelling on her left side of the neck of three-month duration.
Physical examination revealeda single big, fluctuant and painless swelling without defined borders in the left part of the neck, without invasion of the skin the latero-cervical lymph nodes were normal.
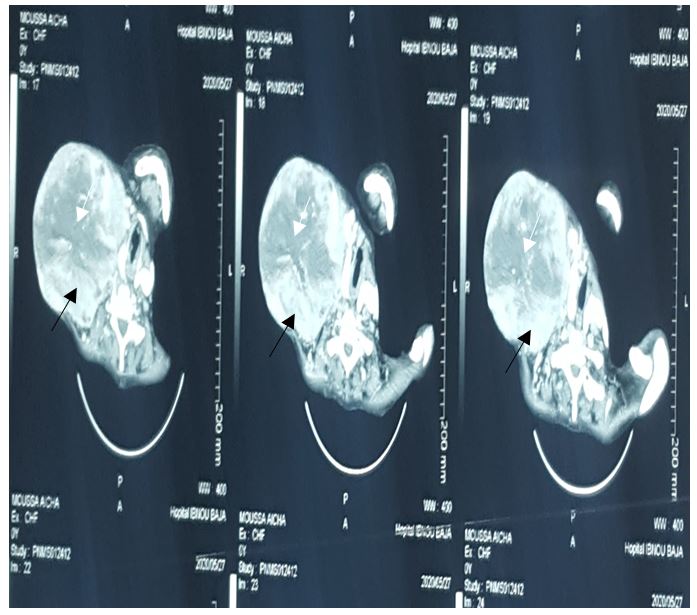
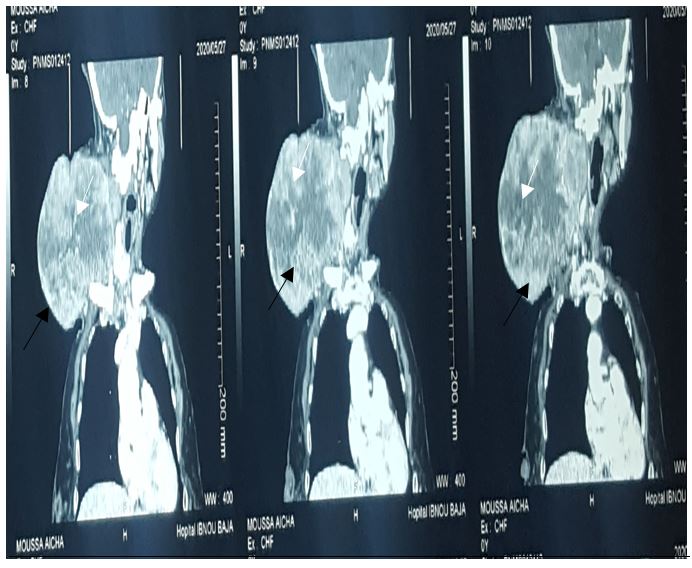
Cervical scan performed detected a big irregular and unlimited tumoral mass measuring approximately 17-13-12 cm with spontaneous central hyperdense area showing intratumor hemorrhage. this mass has heterogeneously enhanced after intravenous injection, with a central neovascularization.
The diagnosis of neurofibroma on neurofibromatosis type 1 was suspected.
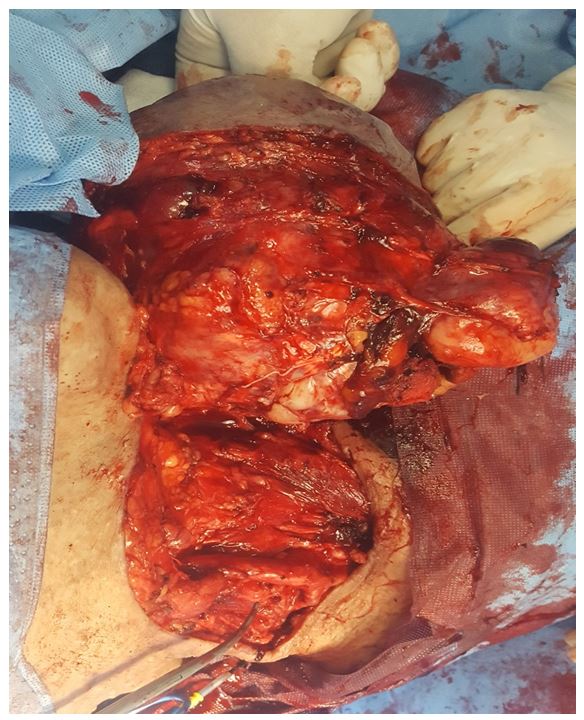
Surgical exploration of our patient found a 16 cm mass of the upper carotid region without vascular nor nerve invasion, and excluded any sign of malignant transformation (no lymphadenopathy nor adhesion to adjacent tissue). Total Excision of the mass with preservation of vascular axis and nervous structure.
The postoperative course was uneventful, and the patient was discharged on the 5th postoperative day in good general condition.
Anatomopathological and immuno-histochemical study confirmed the diagnosis.
At the date, the patient is still without signs of recurrence two years after surgery.
Discussion
Plexiform neurofibroma is a rare,benign and peripheral nerve sheath tumor,it arises from conjunctival perineural cells. [2] it is nearly pathognomonic for neurofibromatosis; this lesion affects 24-32% of NF1 patients [5]. An isolated plexiform neurofibromas without an NF1 is rare [2]
Plexiform neurofibromas carries an increased risk of malignant transformation estimated at 10 – 15% [4]. It is anunencapsulated tumor with infiltrative pattern of surrounding structures: nerve trunks of the different plexus and their dividing branches, adiposetissue and muscle. [2]
The origin of cervical neurofibromas is cranial nerves [1] or the cervical or brachial plexus [6]. Craniofacial localization is rare estimated at 3 to 7% of cases of neurofibromatosis type I,
The excision sacrifices often vascular or nerve structures taken by the tumor. [1]
The diagnosis criteria of NF1 is clinical and is based on the NIH (National Institutes of Health).
Anatomopathological and immuno-histochemical studyconfirmation is essential, especially without suggestive context of neurofibromatosis type I [5]. S100 and CD34 markers are crucial.
Ultrasonography detects a cluster shape mass, hypo-echogenic well limited, oriented along the a nerve trunk, cystic formations as well as posterior reinforcement are observed in 70% of cases. [2]
They can be confused withpathological lymph nodes [5].
The CT scan show nodular, fusiform or cluster images, less dense than muscle (20-30UH) This low density is presence of lipid inclusions in Schwann cells, fat cells, cystic degeneration and a myxoid stroma explains this low density. after intravenous injection, the tumor enhances heterogeneously or homogeneously. [2]
Magnetic resonance imaging sequences is the reference radiological examination, showing a tumoral masshypo signal T1, hyper signal T2 with a pathognomonic central; hypo signal and cockade shape when it is voluminous, enhancement is variable after intravenous injection. [2]
In general, plexiform neurofibroma grow slowly. Rapid growth is observed during puberty or pregnancy without spontaneous regression. Clinical manifestation depends on localization and shape ofthe swelling [2].
For our patient, she presented a rapidly growing mass on the deep spaces of the neck associated to dyspnea and dysphagia.
In the medical literature, isolated cases of NFP mainly concern adults. Aloi and Massobrioen 1989 published the first case of a 35-year-old man with an isolated NFP. Fischer et al. also reported a similar case in 1997. Lin and al. discovered 5 cases out of 51 patients operated on NFP. In 2010, Levy Benchetonand al. published a case of a 35-year-old man with isolated NFP. [5]
Clinically, we suspect malignant progression on the base of rapid growing, induration or painful transformation of the neurofibroma and change of associated pre-existing neurological sign [4]
The surgical excision is the reference treatment.
Neurofibrosarcoma represent the common malignant transformation of NFP; that happens in 3 to 15% of cases.large surgical excision may prevent this risk especially when we suspect clinically malignant transformation. [5]
Large excision with nervous and sometimes vascular sacrifice may be responsible of morbidities. [3]
Surgical exploration of our patient excluded any sign of malignant transformation (no lymphadenopathy nor adhesion to adjacent tissue).
Finally, a multidisciplinary approach including clinical, radiological and surgical practicians and annual monitoring can detect possible recurrence or malignant transformation. [2]
Conclusion
Von Recklinghausen's disease is a common hereditary disease.
Follow-up in specialized multidisciplinary centers allows a rational management. Possible malignant transformation of neurofibromas requires long-term follow-up. The treatment of this pathology is surgical when it is responsible of functional or aesthetic disorders, and anatomopathological examination of the specimen is needed to confirm the diagnosis.
Conflicts of interest
The authors declare no conflict of interest.
- Gamra OB, Romdhane N, Khamassi K, et al. Les tumeursnerveusescervico-faciales : a propos de 47 casExtracranialhead and neck neurogenictumors: report of 47 cases. LA TUNISIE MEDICALE. 2016;94:5.
- Bouimetarhan L, Bellamlih H, En-nafaa I, El Fenni J, Amil T, Radouane B. Neurofibfromeplexiformecervical: à propos d’un cas. Pan Afr Med J. 2018;30. doi:10.11604/pamj.2018.30.41.14446
- Koné FI. NEUROFIBROME GEANT CERVICAL : A PROPOS DE DEUX CAS .Revue Africaine et Malgache de Recherche Scientifique/Sciences de la Santé. 2020;2(1). Accessed May 9, 2021. http://publication.lecames.org/index.php/sante/article/view/1796
- Charfeddine I, Mnejja M, Hammami B, et al. Tumeur maligne des gaines nerveuses périphériques révélant une neurofibromatose type 1. Journal Tunisien d’ORL et de Chirurgie Cervico-Faciale. 2010;20(1). doi:10.4314/jtdorl.v20i1.57940
- Es Seddiki A, Rkain M, Messaoudi S, Benhadou H, Benajiba N. Une cause rare de masse cervicale chez l’enfant : le neurofibrome plexiforme isolé. Archives de Pédiatrie. 2015;22(12):1276-1278. doi:10.1016/j.arcped.2015.09.006
- Shimizu H, Yoshihara T, Sakurai H, Nozaki M. Cervical Neurofibroma in a Patient with von Recklinghausen’sDisease. AurisNasus Larynx. 1994;21(4):253-257. doi:10.1016/S0385-8146(12)80090-3